Establishing a coherent mapping of the relationships among all known proteins is crucial for elucidating processes of protein emergence and evolution. Yet the capacity to fully capture relationships of protein similarity is complicated by the nonstraightforward interplay between sequence and structure; indeed, proteins with unrelated sequences can adopt similar structures, and, conversely, proteins with similar or identical sequences can manifest radically different structures. Here, we introduce Contrastive Learning Sequence–Structure (CLSS), a contrastive protein language model (PLM) trained to coembed sequence and structure information in a self-supervised manner, facilitating a holistic representation of protein relatedness. CLSS represents the structures and sequences of full domains and domain subsequences as vectors in the same high-dimensional latent space. We show that this approach yields meaningful shared representations, which recapitulate the extensive structure- and sequence-based knowledge encoded in human-curated hierarchical protein classification systems (ECOD and CATH). Moreover, the representations generated by CLSS outperform those generated by alternative state-of-the-art PLMs in downstream classification tasks. Notably, we show that even the far larger space of domain subsequences is successfully coembedded, establishing a PLM tailored to these evolutionarily meaningful objects. CLSS embeddings produce informative representations of the protein universe without further downstream processing, as we demonstrate by analyzing preferential associations between protein architectures and ligand types across protein space.


Recent News
Highlights
- 2026/08/04Manuscript accepted to PNAS
- 2026/06/19Manuscript accepted to Journal of Physical Chemistry C
- 2026/04/16Manuscript accepted to Trends in Chemistry
- 2026/01/08Manuscript accepted to Nature Communications
- 2025/08/15Manuscript accepted to Protein Science
- 2025/07/30Manuscript accepted to Bioinformatics Advances
- 2025/06/13Longo Lab Awarded Grant by NASA
Early in the history of life, mineral surfaces, such as the NiFeS mineral violarite (FeNi2S4), may have been the primary catalysts. How these primitive chemical systems could have undergone robust chemical evolution remains unknown. Here, we show that simple amino acids, such as diaminobutyric acid (DAB), can accelerate H+ and/or CO2 electrochemical reduction on violarite. Reactions performed with structurally similar compounds demonstrate that both amino groups of DAB participate in HER catalytic enhancement. Spectroscopic analyses suggest that the role of DAB is to deliver protons to the mineral surface. The observation that simple basic amino acids can enhance surface-supported electrochemistry reveals that amino acids could have produced a positive feedback that promoted metabolic complexification even before their eventual incorporation into complex peptide or protein catalysts.
Proteins make complex life possible, yet our understanding of their emergence remains limited. What are the informational limits of protein folding, and how did the first proteins emerge? Protein simplification studies—in which contemporary folds are built from limited alphabets, symmetrized, fragmented, or shortened—have provided key insights into these questions. These studies use design constraints to address the discoverability of, and connectedness between, protein folds. By considering various environments, such as high salt concentrations or peptide–nucleic acid coacervates, the role of context in the emergence of folded domains is explored. Taken together, these studies support the early emergence of protein folds and reveal the existence of highly connected and readily traversable regions of sequence–structure space.
Polyphosphate kinases (PPKs) catalyze phosphoryl transfer between polyphosphates and nucleotides. Polyphosphates are a cost-effective source of phosphorylating power, making PPKs attractive enzymes for nucleotide production. However, at present, applications that require the simultaneous utilization of diverse nucleotides are not possible due to the restricted substrate profiles of PPKs. Here, we present a universal PPK capable of efficiently phosphorylating all eight common ribonucleotides (purines and pyrimidines, monophosphates and diphosphates) to triphosphates. Under optimal conditions, ~70% triphosphate conversion was observed for each substrate. To demonstrate the biotechnological potential of a universal PPK, we developed a one-pot assay for PPK-powered in vitro transcription. Primitive biology likely relied on enzyme promiscuity to support nascent metabolism with a compact proteome. This work highlights how applying the same principle to synthetic biology can facilitate the construction of complex in vitro reaction systems.
Recent evidence suggests that peptide-RNA coacervates may have buffered the emergence of folded domains from flexible peptides. As primitive peptides were likely composed of both L- and D-amino acids, we hypothesized that coacervates may have also supported the emergence of chiral control. To test this hypothesis, we compared the coacervation propensities of an isotactic (homochiral) peptide and a syndiotactic (alternating chirality) peptide, both with an identical sequence derived from the ancient helix-hairpin-helix (HhH) motif. Using electron paramagnetic resonance (EPR) spectroscopy and atomistic molecular dynamics (MD) simulations, we found that the syndiotactic peptide does not form stable dimers with high α-helicity in solution, unlike the isotactic peptide. However, both peptides do coacervate with RNA, albeit with distinct reentrant phase behaviors. Coacervation in each case is facilitated by oligomer formation, likely dimerization, upon RNA binding that promotes RNA cross-linking. Additionally, RNA cross-linking and coacervation of the syndiotactic peptide may involve α-helical conformations, according to atomistic MD simulations. Coarse-grained MD simulations indicate that the differences in reentrant phase behavior of isotactic and syndiotactic peptides are associated with differences in dimer flexibility and stability, which modulate the strength of peptide-peptide and peptide-RNA interactions and, consequently, the effectiveness of RNA cross-linking. These results illustrate how RNA binding and/or coacervation by early proteins could have promoted the transition of flexible, heterochiral peptides into folded, homochiral domains.
The protein-coding regions of eukaryotic genes are fragmented into exons that, like the genes within which they are situated, can be duplicated, deleted, or reorganized. Cataloging and organizing within-gene exon similarities is necessary for a systematic study of exon evolution and its consequences. To facilitate the study of exon duplications, we present Exonize, a computational tool that identifies and classifies coding exon duplications in annotated genomes. Exonize implements a graph-based framework to handle clusters of related exons resulting from repeated rounds of exon duplication. The interdependence between duplicated exons or groups of exons across transcripts is classified. By identifying duplication events between exonic and intronic regions, Exonize can detect unannotated or degenerate exons. To aid in data parsing and downstream analysis, the Python module exonize_analysis is provided. The application of Exonize to 20 eukaryote genomes identifies full-exon duplications in at least 4% of vertebrate genes, with more than 900 human genes having a full-exon duplication event.
If you are an experimentalist interested in studying protein histories as a graduate student in the Longo Lab at ELSI, please get in touch!
Primitive nucleic acids and peptides likely collaborated in early biochemistry. What forces drove their interactions and how did these forces shape the properties of primitive complexes? We investigated how two model primordial polypeptides associate with DNA. When peptides were coupled to a ferromagnetic substrate, DNA binding depended on the substrate’s magnetic moment orientation. Reversing the magnetic field nearly abolished binding despite complementary charges. Inverting the peptide chirality or just the cysteine residue reversed this effect. These results are attributed to the chiral-induced spin selectivity (CISS) effect, where molecular chirality and electron spin alter a protein’s electric polarizability. The presence of CISS in simple protein–DNA complexes suggests that it played a significant role in ancient biomolecular interactions. A major consequence of CISS is enhancement of the kinetic stability of protein–nucleic acid complexes. These findings reveal how chirality and spin influence bioassociation, offering insights into primitive biochemical evolution and shaping contemporary protein functions.
The helix-hairpin-helix (HhH) motif is an ancient and ubiquitous nucleic acid-binding element that has emerged as a model system for studying the evolution of dsDNA-binding domains from simple peptides that phase separate with RNA. We analyzed the entire putative evolutionary trajectory of the HhH motif – from a flexible peptide to a folded domain – for functional robustness to total chiral inversion. Against expectations, functional ‘ambidexterity’ was observed for both the phase separation of HhH peptides with RNA and binding of the duplicated (HhH)2-Fold to dsDNA. Moreover, dissociation kinetics, mutational analysis, and molecular dynamics simulations revealed overlap between the binding modes adopted by the natural and mirror-image proteins to natural dsDNA. The similarity of several dissociation phases upon chiral inversion may reflect the history of (HhH)2-Fold binding, with the ultimate emergence of a high-affinity binding mode, supported by a bridging metal ion, depopulating but not displacing more primitive (potentially ambidextrous) modes. These data underscore the surprising functional robustness of the HhH protein family and suggest that the veil between worlds with alternative chiral preferences may not be as impenetrable as is often assumed.
If you are an experimentalist interested in studying protein histories as a graduate student in the Longo Lab at ELSI, please get in touch!
At the heart of many nucleoside triphosphatases is a conserved phosphate-binding sequence motif. A current model of early enzyme evolution proposes that this 6-8 residue motif could have sparked the emergence of the very first nucleoside triphosphatases – a striking example of evolutionary continuity from simple beginnings, if true. To test this provocative model, seven disembodied Walker A-derived peptides were extensively computationally characterized. Although dynamic flickers of nest-like conformations were observed, significant structural similarity between the situated peptide and its disembodied counterpart was not detected. Simulations suggest that phosphate binding is non-specific, with a preference for GTP over orthophosphate. Control peptides with the same amino acid composition but different sequences and situated conformations behaved similarly to the Walker A peptides, revealing no indication that the Walker A sequence is privileged as a disembodied peptide. We conclude that the evolutionary history of the P-Loop NTPase family is unlikely to have started with a disembodied Walker A peptide in an aqueous environment. The limits of evolutionary continuity for this protein family must be reconsidered. Finally, we argue that motifs such as the Walker A motif may represent incomplete or fragmentary molecular fossils – the true nature of which have been eroded by time.
Polyphosphate kinase 2 (PPK2) enzymes catalyze phosphoryl transfer from polyphosphate to nucleotides and are divided into three classes, each presumed to have different catalytic preferences. With relevance to biotechnology, medicine, and primitive biology, there is significant interest in understanding the evolutionary history of PPK2 enzymes and predicting their functional properties. We reasoned that the distribution and pairing preferences of PPK2 gene classes across the prokaryote tree of life may shed light on these questions. PPK2 was found to be a dynamic gene family, often present in only a subset of species within a clade, even when considering a single genus. Although all possible PPK2 pairs were observed, a ~2-fold enrichment for Class I enzymes in species with multiple PPK2 genes strongly shapes pairing preferences. PPK2 class preference in the absence of PPK1, which synthesizes rather than utilizes polyphosphate, indicates the potential for functional adaptation and/or promiscuity with respect to reaction directionality for all classes, a feature that has previously been associated only with Class I. Patterns of adjacent PPK2 genes revealed signatures of gene duplication, as adjacent genes overwhelmingly belonged to the same class, as well as the potential for an added layer of PPK2 dynamics: hetero-oligomerization of single-domain Class II enzymes to recapitulate the structure of two-domain Class II enzymes. Finally, an updated PPK2 tree constructed from domains instead of genes calls into question established narratives of PPK2 evolution, putting new limits on the extent to which nucleobase promiscuity can be invoked in the early evolution of this family.
In 1966, Margaret Dayhoff reasoned that duplication and fusion is a fundamental mechanism for generating protein complexity. Her insight inspired generations of scientists, several of whom would demonstrate this trajectory with short peptides that symmetrically assemble into a contemporary protein architecture. But how did these oligomerizing peptides, able to adopt complex conformations, emerge in the first place? In the present review, the evolution of an ancient and ubiquitous nucleic acid-binding element is traced from a simple, heterochiral peptide that coacervates with RNA to a folded-domain that binds with high affinity to the minor groove of double-stranded DNA.
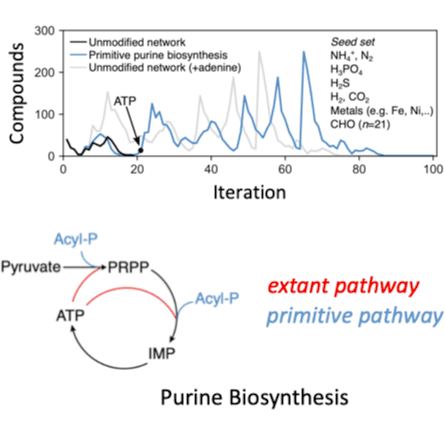
An unresolved question in the origin and evolution of life is whether a continuous path from geochemical precursors to the majority of molecules in the biosphere can be reconstructed from modern-day biochemistry. Here we identified a feasible path by simulating the evolution of biosphere-scale metabolism, using only known biochemical reactions and models of primitive coenzymes. We find that purine synthesis constitutes a bottleneck for metabolic expansion, which can be alleviated by non-autocatalytic phosphoryl coupling agents. Early phases of the expansion are enriched with enzymes that are metal dependent and structurally symmetric, supporting models of early biochemical evolution. This expansion trajectory suggests distinct hypotheses regarding the tempo, mode and timing of metabolic pathway evolution, including a late appearance of methane metabolisms and oxygenic photosynthesis consistent with the geochemical record. The concordance between biological and geological analyses suggests that this trajectory provides a plausible evolutionary history for the vast majority of core biochemistry

Understanding the emergence and evolution of agency is one of the great challenges in both biological research and philosophy. Hiroki Kojima (University of Tokyo), Eran Agmon (University of Connecticut), and I will bring agential studies into the laboratory by constructing a real-time evolution platform in which populations of organic (B-life) and digital (A-life) organisms are under mutual control and poised to form mutualistic community structures. Will community structure promote the emergence of agential features in the A-life population? Stay tuned! Calls for two open positions in Tokyo (Longo Lab) and one open position in Connecticut (Agmon Lab with support mentorship from Co-I Kojima) will follow shortly.

After a brief stint at her home institution in Stockholm, Marina has returned to the Longo Lab to continue her research on the evolution of exon structure (and provide some much needed programming mentorship to all those around her). Welcome back, Marina!

Prof. Gruić Sovulj is an expert in the enzymology of amino-acyl tRNA synthetases, the enzymes that define the genetic code. During her 2-month visit to the Longo Lab at ELSI, she will use bioinformatic approaches to study the evolution of this most important enzyme class.

Tatsuya received a Bachelor's degree in Biological Chemistry from the University of Toronto. As a student in the ELSI Graduate Program, his research efforts will focus on the origin of life and the emergence of complex biopolymer structure. Welcome, Tatsuya!

My passion for science started with Science Fair, and so it was a great pleasure to be able to share my research on early protein evolution with these amazingly talented young scientists! Image credit: ELSI
Marina (left) is a Mathematics PhD student working in the laboratory of Lars Arvestad where she studies the evolution of exon duplication. At ELSI, she will explore the consequences of exon duplication from the perspective of protein structure evolution.
Sarah (right) is a Bioinformatics PhD student working in the laboratory of Peter Stadler where she develops methods for RNA structure prediction. At ELSI, she will study the forces that drive protein sequence convergence between distantly related species.
Nat/Ivy is a diverse and ubiquitous CoA-binding evolutionary lineage that catalyzes acyltransferase reactions, primarily converting thioesters into amides. At the heart of the Nat/Ivy fold is a phosphate-binding loop that bears a striking resemblance to that of P-loop NTPases – both are extended, glycine-rich loops situated between a β-strand and an α-helix. Nat/Ivy, therefore, represents an intriguing intersection between thioester chemistry, a putative primitive energy currency, and an ancient mode of phospho-ligand binding. Current evidence suggests that Nat/Ivy emerged independently of other cofactor-utilizing enzymes, and that the observed structural similarity – particularly of the cofactor binding site – is the product of shared constraints instead of shared ancestry. The reliance of Nat/Ivy on a β-α-β motif for CoA binding highlights the extent to which this simple structural motif may have been a fundamental evolutionary ‘nucleus’ around which modern cofactor-binding domains condensed, as has been suggested for HUP domains, Rossmanns, and P-loop NTPases. Finally, by dissecting the patterns of conserved interactions between Nat/Ivy families and CoA, the coevolution of the enzyme and the cofactor was analyzed. As with the Rossmann, it appears that the pyrophosphate moiety at the center of the cofactor predates the enzyme, suggesting that Nat/Ivy emerged sometime after the metabolite dephospho-CoA.

Stunning views and exciting talks at the Annual Meeting of the Biophysical Society of Japan. Check out my poster, describing a project in collaboration with the Prof. Norman Metanis and Prof. Koby Levy, here!

Although OIST is surrounded by stunning beaches, the real draw is the cutting-edge research happening on campus. Thanks so much to fellow Tawfik Lab alumnus Prof. Paola Laurino for inviting me!
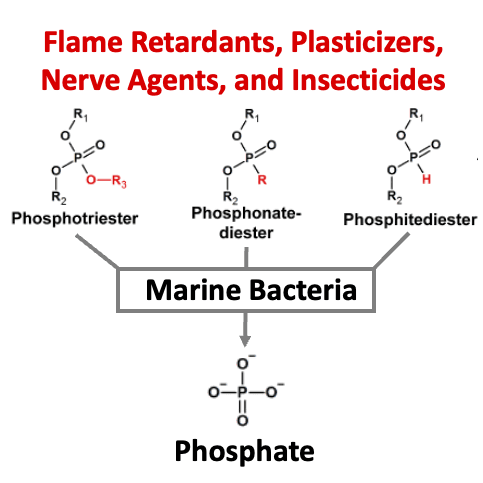
Anthropogenic organophosphorus compounds (AOPCs), such as phosphotriesters, are used extensively as plasticizers, flame retardants, nerve agents, and pesticides. To date, only a handful of soil bacteria bearing a phosphotriesterase (PTE), the key enzyme in the AOPC degradation pathway, have been identified. Therefore, the extent to which bacteria are capable of utilizing AOPCs as a phosphorus source, and how widespread this adaptation may be, remains unclear. Marine environments with phosphorus limitation and increasing levels of pollution by AOPCs may drive the emergence of PTE activity. Here, we report the utilization of diverse AOPCs by four model marine bacteria and 17 bacterial isolates from the Mediterranean Sea and the Red Sea. To unravel the details of AOPC utilization, two PTEs from marine bacteria were isolated and characterized, with one of the enzymes belonging to a protein family that, to our knowledge, has never before been associated with PTE activity. When expressed in Escherichia coli with a phosphodiesterase, a PTE isolated from a marine bacterium enabled growth on a pesticide analog as the sole phosphorus source. Utilization of AOPCs may provide bacteria a source of phosphorus in depleted environments and offers a prospect for the bioremediation of a pervasive class of anthropogenic pollutants.
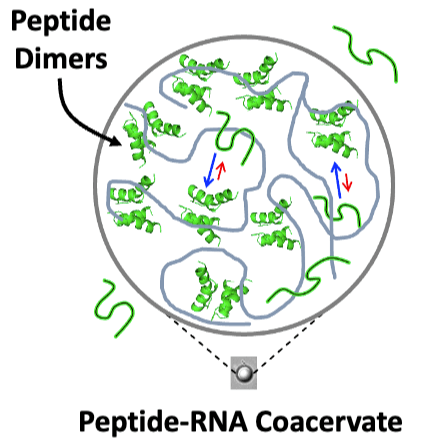
Peptide-RNA coacervates can result in the concentration and compartmentalization of simple biopolymers. Given their primordial relevance, peptide-RNA coacervates may have also been a key site of early protein evolution. However, the extent to which such coacervates might promote or suppress the exploration of novel peptide conformations is fundamentally unknown. To this end, we used electron paramagnetic resonance spectroscopy (EPR) to characterize the structure and dynamics of an ancient and ubiquitous nucleic acid binding element, the helix-hairpin-helix (HhH) motif, alone and in the presence of RNA, with which it forms coacervates. Double electron–electron resonance (DEER) spectroscopy applied to singly labeled peptides containing one HhH motif revealed the presence of dimers, even in the absence of RNA. Moreover, dimer formation is promoted upon RNA binding and was detectable within peptide-RNA coacervates. DEER measurements of spin-diluted, doubly labeled peptides in solution indicated transient α-helical character. The distance distributions between spin labels in the dimer and the signatures of α-helical folding are consistent with the symmetric (HhH)2-Fold, which is generated upon duplication and fusion of a single HhH motif and traditionally associated with dsDNA binding. These results support the hypothesis that coacervates are a unique testing ground for peptide oligomerization and that phase-separating peptides could have been a resource for the construction of complex protein structures via common evolutionary processes, such as duplication and fusion.

The Blue Marble Space Institute of Science (BMSIS) is an international group of scientists interested in basic science, sustainability, and public engagement. I can't wait to start learning together with the kindred sprints at BMSIS!
At the IdeasLab, I had the opportunity to work alongside computer scientists and physicists as we developed projects to probe the nature of mind, agency, and the process of abiogenesis. Many thanks to the John Templeton Foundation for making this trip possible -- I learned so much from my peers and mentors, and it's a week I won't soon forget!
Check out made-for-YouTube versions of the talks here. As these papers are not yet published, the stories are subject to change! A collaboration between my lab and the Metanis Lab (Hebrew University of Jerusalem) about "ambidextrous" protein folds: A peek at some conversations between me and my former Postdoctoral Advisor Shawn McGlynn:

I've been promoted to Specially Appointed Associate Professor at the Earth-Life Science Institute (ELSI) at the Tokyo Institute of Technology! If you are interested protein evolution or metabolism and would like to join the Longo Lab @ ELSI (Master's or PhD level), please drop me a line!
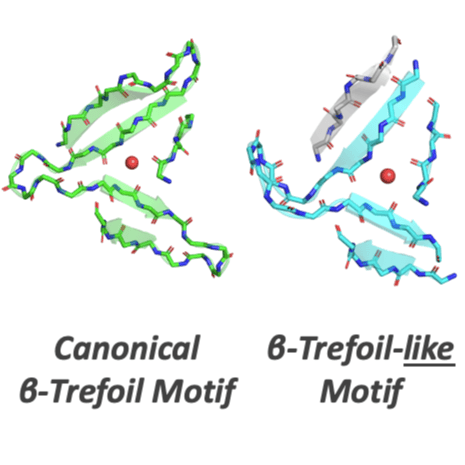
As sequence and structure comparison algorithms gain sensitivity, the intrinsic interconnectedness of the protein universe has become increasingly apparent. Despite this general trend, β-trefoils have emerged as an uncommon counterexample: They are an isolated protein lineage for which few, if any, sequence or structure associations to other lineages have been identified. If β-trefoils are, in fact, remote islands in sequence-structure space, it implies that the oligomerizing peptide that founded the β-trefoil lineage itself arose de novo. To better understand β-trefoil evolution, and to probe the limits of fragment sharing across the protein universe, we identified both ‘β-trefoil bridging themes’ (evolutionarily-related sequence segments) and ‘β-trefoil-like motifs’ (structure motifs with a hallmark feature of the β-trefoil architecture) in multiple, ostensibly unrelated, protein lineages. The success of the present approach stems, in part, from considering β-trefoil sequence segments or structure motifs rather than the β-trefoil architecture as a whole, as has been done previously. The newly uncovered inter-lineage connections presented here suggest a novel hypothesis about the origins of the β-trefoil fold itself – namely, that it is a derived fold formed by ‘budding’ from an Immunoglobulin-like β-sandwich protein. These results demonstrate how the evolution of a folded domain from a peptide need not be a signature of antiquity and underpin an emerging truth: few protein lineages escape nature’s sewing table.

We are deeply saddened by the premature and unexpected passing of our dear friend and mentor Dan Salah Tawfik (דן סלח תופיק). Danny’s contributions to science were immense, and his insights into enzyme catalysis have shaped an array of fields. In this obituary, we try to communicate the impact Danny had on us personally as a mentor, and the joy of doing science in Danny's lab. Photo by David Salem of Zoog Productions.
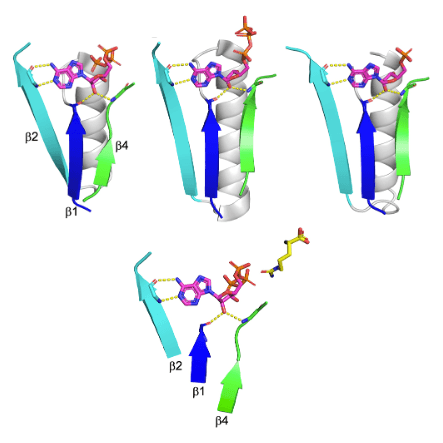
Among the enzyme lineages that undoubtedly emerged prior to the last universal common ancestor is the so-called HUP, which includes Class I aminoacyl tRNA synthetases (AARSs) as well as enzymes mediating NAD, FAD, and CoA biosynthesis. Here, we provide a detailed analysis of HUP evolution, from emergence to structural and functional diversification. The HUP is a nucleotide binding domain that uniquely catalyzes adenylation via the release of pyrophosphate. In contrast to other ancient nucleotide binding domains with the aba sandwich architecture, such as P-loop NTPases, the HUP’s most conserved feature is not phosphate binding, but rather ribose binding by backbone interactions to the tips of b1 and/or b4. Indeed, the HUP exhibits unusual evolutionary plasticity and, while ribose binding is conserved, the location and mode of binding to the base and phosphate moieties of the nucleotide, and to the substrate(s) reacting with it, have diverged with time, foremost along the emergence of the AARSs. The HUP also beautifully demonstrates how a well-packed scaffold com- bined with evolvable surface elements promotes evolutionary innovation. Finally, we offer a scenario for the emergence of the HUP from a seed bab fragment, and suggest that despite an identical architecture, the HUP and the Rossmann represent independent emergences.
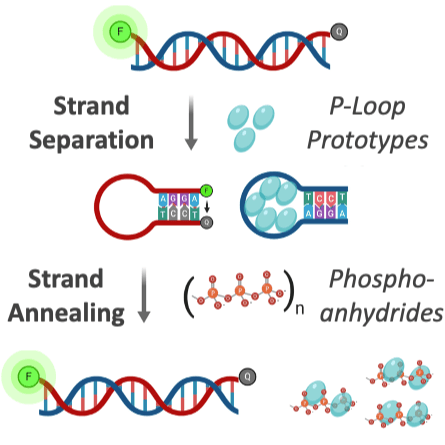
It is widely assumed that today’s large and complex proteins emerged from much shorter and simpler polypeptides. Yet the nature of these early precursors remains enigmatic. We describe polypeptides that contain one of the earliest protein motifs, a phosphate-binding loop, or P-loop, embedded in a single beta-alpha element. These P-loop prototypes show intriguing characteristics of a primordial world comprised of nucleic acids and peptides. They are ‘generalists’ capable of binding different phospho-ligands, including inorganic polyphosphates and single-stranded DNA. Nonetheless, in promoting double-stranded DNA unwinding and strand-exchange they resemble modern P-loop helicases and recombinases. Our study describes a missing link in the evolution of complex proteins – simple polypeptides that tangibly relate to contemporary P-loop enzymes in sequence, structure and function.
Dating back to the last universal common ancestor (LUCA), the P-loop NTPases and Rossmanns now comprise the most ubiquitous and diverse enzyme lineages. Intriguing similarities in their overall architecture and phosphate binding motifs suggest common ancestry; however, due to a lack of sequence identity and some fundamental structural differences, these families are considered independent emergences. To address this longstanding dichotomy, we systematically searched for ‘bridge proteins’ with structure and sequence elements shared by both lineages. We detected homologous segments that span the first βαβ segment of both lineages and include two key functional motifs: (i) a phosphate binding loop – the ‘Walker A’ motif of P-loop NTPases or the Rossmann equivalent, both residing at the N-terminus of α1; and (ii) an Asp at the tip of β2. The latter comprises the ‘Walker B’ aspartate that chelates the catalytic metal in P-loop NTPases, or the canonical Rossmann β2-Asp that binds the cofactor’s ribose moiety. Tubulin, a Rossmann GTPase, demonstrates the potential of the β2-Asp to take either one of these two roles. We conclude that common P-loops/Rossmann ancestry is plausible, although convergence cannot be completely ruled out. Regardless, both lineages most likely emerged from a polypeptide comprising a βαβ segment carrying the above two functional motifs, a segment that comprises the core of both enzyme families to this very day.
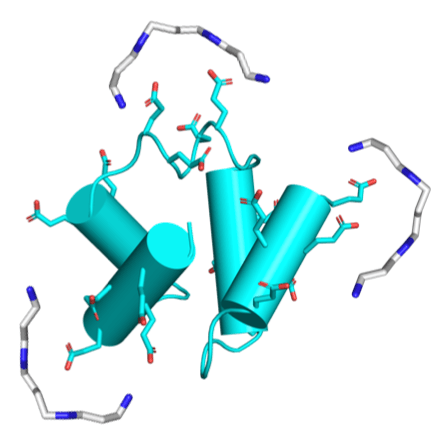
Polyamines are known to mediate diverse biological processes, and specifically to bind and stabilize compact conformations of nucleic acids, acting as chemical chaperones that promote folding by offsetting the repulsive negative charges of the phosphodiester backbone. However, whether and how polyamines modulate the structure and function of proteins remains unclear. Further, early proteins are thought to have been highly acidic, like nucleic acids, due to a scarcity of basic amino acids in the prebiotic context. Perhaps polyamines, the abiotic synthesis of which is simple, could have served as chemical chaperones for such primordial proteins? We replaced all lysines of an ancestral 60-residue helix-bundle protein to glutamate, resulting in a disordered protein with 21 glutamates in total. Polyamines efficiently induce folding of this hyper-acidic protein at sub-millimolar concentrations, and their potency scaled with the number of amine groups. Compared to cations, polyamines were several orders of magnitude more potent than Na+, while Mg2+ and Ca2+ had an effect similar to a di-amine, inducing folding at approximately seawater concentrations. We propose that (i) polyamines and dications may have had a role in promoting folding of early proteins devoid of basic residues, and that (ii) coil-helix transitions could be the basis of polyamine regulation in contemporary proteins.

¿Cómo eran las primeras proteínas de la historia? [La Razón]
Israeli scholars reveal structure of 3.7 billion-year-old proteins [The Jerusalem Post]
A new study hints at how non-living matter coalesced into the first living cells [Salon]
In addition, this article was covered by the This Week in Evolution (TWiEVO) podcast and highlighted by Faculty Opinions as being of exceptional importance!
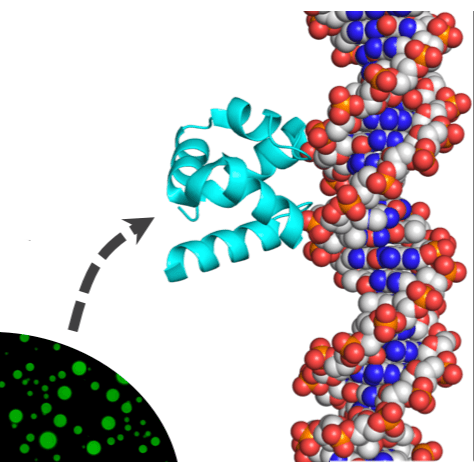
The first proteins emerged some 4 billion years ago, and understanding how they came about is a daunting challenge. Further complicating matters, the rules of protein structure and function derived from modern proteins may be irrelevant to their earliest ancestors. We report an integrated approach in which protein sequence, structure, and function are considered. We show that a simple function (phase separation) may have served as the basis for a complex function (specific double-stranded DNA binding), and that disordered polypeptides can give rise to structured, well-packed domains. Finally, we demonstrate that functional proteins may arise from short and simple sequences that include ornithine, an amino acid likely present in early proteins yet absent in modern proteins.
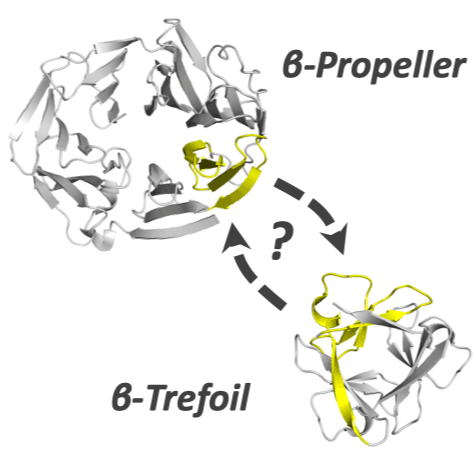
Many protein architectures exhibit evidence of internal rotational symmetry postulated to be the result of gene duplication/fusion events involving a primordial polypeptide motif. A common feature of such structures is a domain-swapped arrangement at the interface of the N- and C-termini motifs and postulated to provide cooperative interactions that promote folding and stability. De novo designed symmetric protein architectures have demonstrated an ability to accommodate circular permutation of the N- and C-termini in the overall architecture; however, the folding requirement of the primordial motif is poorly understood, and tolerance to circular permutation is essentially unknown. The β-trefoil protein fold is a threefold-symmetric architecture where the repeating ~42-mer "trefoil-fold" motif assembles via a domain-swapped arrangement. The trefoil-fold structure in isolation exposes considerable hydrophobic area that is otherwise buried in the intact β-trefoil trimeric assembly. The trefoil-fold sequence is not predicted to adopt the trefoil-fold architecture in ab initio folding studies; rather, the predicted fold is closely related to a compact "blade" motif from the β-propeller architecture. Expression of a trefoil-fold sequence and circular permutants shows that only the wild-type N-terminal motif definition yields an intact β-trefoil trimeric assembly, while permutants yield monomers. The results elucidate the folding requirements of the primordial trefoil-fold motif, and also suggest that this motif may sample a compact conformation that limits hydrophobic residue exposure, contains key trefoil-fold structural features, but is more structurally homologous to a β-propeller blade motif.
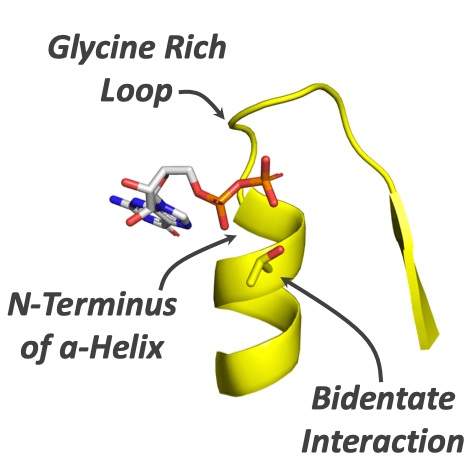
The first enzymes emerged ∼4 billion years ago and have subsequently become the most diverse and functionally important component of life. But what were the first enzymes doing and how did they look? We probed the properties of the first enzymes by analyzing phospho-ligand binding across all known protein evolutionary lineages. We find that phospho-ligand binding was the founding function of the most ancient enzymes. As opposed to younger evolutionary lineages, ancient enzymes preferentially use N termini of α-helices to bind phosphate moieties. The dominance of N-helix binding sites in the earliest enzymes reflects the ability of the α-helix to realize binding via short and simple sequences, including serines and threonines that interact via both the backbone and side chain.
